

The origin of the mysterious, huge and anomalously cold region of the sky, which was called the Superfamily Eridani, for many years remained the subject of debate among scientists. In 2015, researchers declared this region in the constellation Eridan a “super-voodoo” – a space with an extremely low density of galaxies compared to the rest of the universe. However, the result of this study was not reproduced by other scientists.
In a new study, the results of which were published by the Royal Astronomical Society, British scientists from the University of Durham are disputing the theory of super-Vedic forms. At the same time, it is suggested that the relic cold spot can be evidence of a collision of our universe with a certain parallel universe.
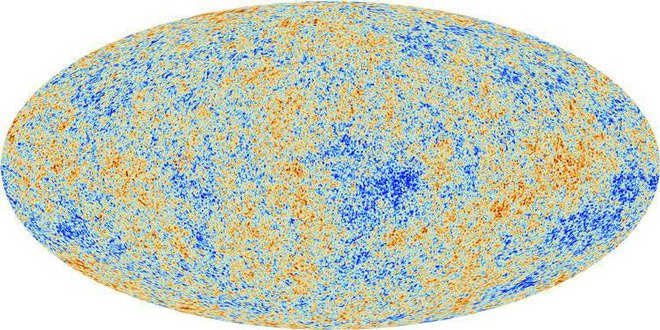
Map of “cosmic microwave background radiation”. © ESA and the Planck Collaboration
A cold spot can be seen on the maps of “cosmic microwave background radiation” (CMB), which is radiation that has survived to our days since the birth of the universe. Scientists say that CMB is a photograph of what the universe looked like when it was 380 thousand years old, and its temperature was 3000 degrees Kelvin. Previous studies have shown that the temperature of the cold spot is approximately 70 μK colder than the average temperature of the entire relic background (with average temperature fluctuations in the CMB – 18 μK).
There are two main theories explaining the nature of the cold spot. The first is the theory of super-waters, through which the light passed. It was also suggested that this is an initially very cold area of the early universe. The authors of the new study attempted to establish the truth by comparing new data on galaxies around a cold spot with data from another region of the starry sky. New data were obtained with the help of an Anglo-Australian telescope operating at the Siding Spring Observatory, the previous ones in the framework of the GAMA project on the study of galaxies and mass clusters.
In the framework of the GAMA study and similar ones, the “spectra” of thousands of galaxies are recorded. These spectra are images of light emanating from the galaxy and propagating in accordance with its wavelength. So scientists get an idea of the structure of those streams that are radiated by various elements in a particular galaxy. The further the galaxy is located, the greater the expansion of the Universe shifts these lines to longer waves. Due to the so-called “red shift” scientists can calculate the distance to the galaxy. Spectra in combination with the position on the starry sky allow you to build three-dimensional maps of the distribution of galaxies.
However, the researchers concluded that to explain the cold spot is simply not enough galaxies – nothing special about the distribution of galaxies in front of the cold spot, compared with other areas of the starry sky, scientists have not found.
From this, the researchers conclude that if the cold spot was not caused by a super-void, then, most likely, it is a very large cold area, from which the relic microwave background radiation originated. One of the most exotic explanations of its source today is the collision of universes at a very early stage.

Galaxies on the expanses of the universe. © Noodle snacks
The idea that we live in a “multiverse” consisting of an infinite number of parallel universes has long been not excluded by scientists. However, until now, physicists have not agreed on whether the multiverse can be a physical reality or just a bizarre mathematical concept. This is a consequence of such important theories as quantum mechanics, string theory and the theory of inflation of the universe.
Thus, adherents of quantum mechanics claim that any particle can exist in a “superposition”, that is, be in several different states simultaneously (for example, in different places). This statement was experimentally confirmed. However, even with mathematical arguments in favor of the theory of the existence of parallel universes, scientists are still in no hurry to unequivocally recognize the cold spot as evidence of a collision between two universes.
So, Professor of Astrophysics Ivan Baldry of Liverpool John Moors University in his article on the portal The Conversation calls such a probability, voiced by his colleagues, extremely unlikely.
“There is no specific reason why we would now see the imprint of a collision of universes. From what we know about how the universe was being formed, it seems likely that it is much larger than we can see. Therefore, even if parallel universes exist, and we are faced with one of them – which in itself is unlikely – the chances that we will be able to see it in that part of the universe that we can observe in the sky are amazingly small, “says Ivan Baldri.
Meanwhile, another explanation for the nature of Eridan Superspot can be natural fluctuations in the mass density, which lead to temperature changes in the CMB.
Thus, Ivan Baldri sums up, the cold spot is still a mystery for scientists and requires detailed study.









